
Obesity disrupts mitochondria, reduces fat-burning
At a Glance
- Scientists identified a protein that triggers changes in mitochondria that lead to reduced fat-burning to promote weight gain in mice fed a high-fat diet.
- The findings may point to potential new avenues for treating or preventing obesity in people.
Adipose tissue, or body fat, plays a key role in maintaining our health. It helps to store and supply energy, regulate body temperature, and send hormone signals that affect many body functions. But when a person develops obesity, it leads to expansion of a type of fat called white adipose tissue, along with increased inflammation and metabolic changes.
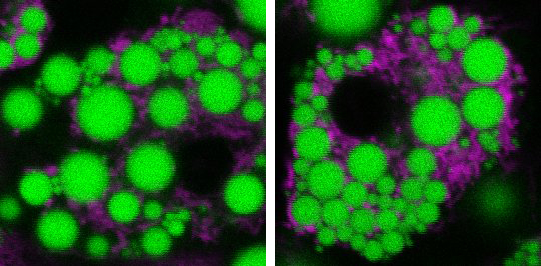
Mitochondria, the energy-generating structures found within cells, are dynamic—that is, they can fuse, change shape, and divide. These changes affect how much energy mitochondria can burn. Some studies have found that obesity can alter these dynamics and cause mitochondria to fragment, making it more difficult for fat cells to burn energy. This might help explain why it can be hard for people with obesity to lose weight. The breakdown of mitochondria has also been tied to insulin resistance in obesity. And insulin resistance is associated with diabetes and other metabolic conditions. But the underlying connections between obesity, mitochondria, and white fat have been unclear.
A research team led by Dr. Alan Saltiel at the University of California, San Diego, had previously shown that a protein called RalA could be activated by insulin in fat cells and promote glucose uptake by brown fat. The team suspected that study of RalA might also give insights into the mitochondrial changes linked to obesity.
To investigate, the researchers fed mice a high-fat diet (about 60% fat) for 8 to 12 weeks. They then examined its effects on RalA and the mitochondria in fat cells. Results were reported on January 29, 2024, in Nature Metabolism.
The researchers found that the high-fat diet caused the mitochondria in white fat tissue to divide into many smaller pieces. As a result, the mitochondria became less effective at burning energy. The high-fat diet also boosted levels of RalA in white fat. These changes weren’t seen in brown fat tissue. The results hinted that higher RalA activity might be the culprit behind many metabolic problems seen in obesity.
To gain insights into the effects of RalA activation, the team looked at what happens in the protein’s absence. They created genetically altered mice that lacked the RalA-producing gene in fat tissues. When given a high-fat diet, mice without RalA in their white fat tissue were protected against diet-induced weight gain and obesity. They had additional metabolic improvements as well. These included better liver function and glucose tolerance, and energy expenditure similar to mice fed a regular diet. Absence of RalA also prevented fragmentation of mitochondria in mice fed a high-fat-diet, thereby protecting mitochondria’s fat-burning functions.
Further analysis showed how RalA activity leads to changes in mitochondria dynamics. The researchers found the same mechanisms in white fat from people. They also found that the activity of a key protein in the process was associated with human obesity. More study will be needed to understand how a high-fat diet raises levels of RalA in white fat in the first place.
“In essence, chronic activation of RalA appears to play a critical role in suppressing energy expenditure in obese adipose tissue,” Saltiel says. “By understanding this mechanism, we’re one step closer to developing targeted therapies that could address weight gain and associated metabolic dysfunctions by increasing fat burning.”
—by Vicki Contie
References: Obesity causes mitochondrial fragmentation and dysfunction in white adipocytes due to RalA activation. Xia W, Veeragandham P, Cao Y, Xu Y, Rhyne TE, Qian J, Hung CW, Zhao P, Jones Y, Gao H, Liddle C, Yu RT, Downes M, Evans RM, Rydén M, Wabitsch M, Wang Z, Hakozaki H, Schöneberg J, Reilly SM, Huang J, Saltiel AR. Nat Metab. 2024 Jan 29. doi: 10.1038/s42255-024-00978-0. Online ahead of print. PMID: 38286821.
Funding: NIH’s National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), Office of the Director (OD), and National Cancer Institute (NCI); American Diabetes Association; Karolinska Institute; NOMIS Foundation; Larry L. Hillblom Foundation; Margareta of Uggla’s foundation; Swedish Research Council; Novo Nordisk Foundation; Knut and Alice Wallenberg’s Foundation; Swedish Diabetes Foundation; Stockholm County Council.






